
The Food and Drug Administration (FDA)’s Circulatory System Devices Panel of the Medical Devices Advisory has made recommendations for leadless pacemakers regarding adverse events, long-term safety issues (including battery longevity), necessary elements for postmarket surveillance, indications for use and labelling, and implanting physicians’ training.
These recommendations came after the Panel, chaired by Richard Page (University of Wisconsin, School of Medicine and Public Health, Madison, USA) and including 16 more members (see the complete list of members below) met on 18 February 2016 in Gaithersburg, USA.
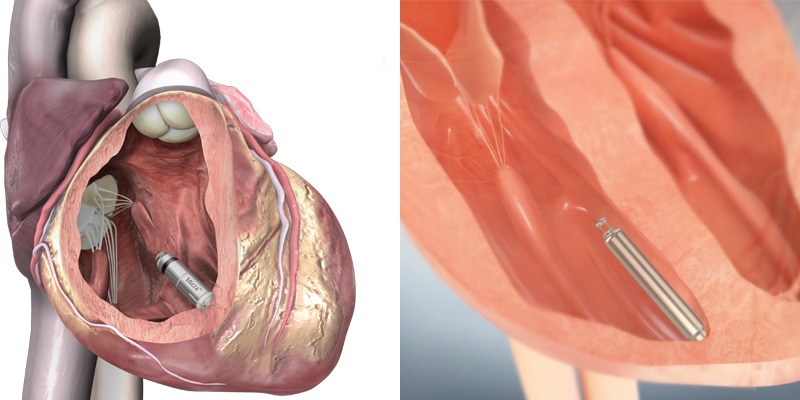
At the beginning of the meeting, the FDA presented an executive summary memorandum on general issues of leadless pacemaker devices. In the summary, the FDA described the current available leadless pacemakers (Micra from Medtronic and Nanostim from St Jude Medical), which are being studied in the USA for VVIR pacing, and presented publicly available data on clinical trials for these devices. The FDA also mentioned that Boston Scientific recently announced plans to develop a pacemaker to complement its Subcutaneous Implantable Cardioverter Defibrillator (S-ICD).
In the summary, knowledge gaps regarding intermediate and long-term performance were addressed. The FDA stated: “The long term (5-10 years) safety profile of the leadless devices is currently unknown. On this issue, the FDA is not aware of late device failure rates or late (greater than two years) complications. Additionally, long-term effectiveness and overall battery longevity in the real-world setting are unknown. The FDA is only aware of battery projections based on bench testing, which suggest that with projected use of 1.5 volts at a pulse width of 0.24 miliseconds, there should be reasonable battery longevity of 12.5 years… It is also currently unclear how the leadless devices should be handled at their end of life. Substantial data on encapsulation, explanation and co-implantation are lacking and the FDA is considering best approaches to collect data to determine how best to resolve these key end of life issues.”
Later in the meeting, representatives from Medtronic and St Jude Medical discussed trial details, plans for post approval studies and implanting physician training programmes.
FDA questions to Panel
At the meeting, the Panel was asked four general questions regarding adverse events, design of post approval studies, appropriate labelling and indications for use. A brief summary from the meeting with the recommendations from the Panel was thereafter released by the FDA.
Adverse events and training
The first question sought to investigate the occurrence of adverse events observed to occur at implant with leadless pacemaker devices as compared to traditional pacemakers. In the brief summary of the Panel meeting it was reported that the Panel felt cardiac perforation rates for leadless pacemakers “should be consistent” with published perforation rates of transvenous pacemakers. Additionally, the panel raised concerns about other adverse events such as pericardial effusion, dislodgement, embolisation, serious groin complications necessitating repair or transfusions, cardiac mortality and infection.
In the brief summary, it was also determined that no subgroup of patients “should be excluded from receiving this device.” It was also highlighted and agreed that implanting physicians “must be adequately trained/informed regarding adverse events and appropriate patient selection.”
Post approval studies design
The second question explored methods of best data collection for acute performance/implant experience in the post approval setting. In the brief summary, the Panel agreed that the acute events should be captured through collection of post approval data, including groin complications, haematoma, vascular issues, perforations, infection, acute electrical performance, and mortality. According to the Panel, the sample size needed to capture acute complications should be around 1,741 patients enrolled. This assumes a complication rate of 1% with a confidence interval width of ±0.5%.
In the brief summary it was also noted that the Panel agreed that, based on the current post approval studies paradigm for cardiac leads, a complication-free rate (including deaths) can be used as the endpoint for long-term performance assessment of leadless pacemakers. The Panel also determined that eight to nine years of follow-up would be necessary for leadless pacemakers.
Regarding scenarios for end of life devices, the Panel suggested collection of observational data when explant of the leadless pacemaker occurs and implant of another leadless pacemaker or a transvenous pacemaker system is performed or an implantable cardioverter defibrillator-in the event the patient develops an indication for one. Another scenario is turning off the existing leadless pacemaker and implant and adjacent leadless pacemaker or adjacent transvenous pacemaker or adjacent ICD.
Post approval studies should also include data on device removal and extraction experience including times it is attempted, success rates and associated complications. Additionally, these studies should incorporate data collection for patients who receive a leadless pacemaker as a replacement for a transvenous system, the brief summary reported.
Labelling
The third question explored appropriate labelling for leadless pacemakers. The Panel confirmed that labelling “should be device-specific and incorporate all experience on long-term performance and end of life options to date, noting limitation of data available at the time of approval,” according to the brief summary.
Indications for use
The last question enquired about the language and type of information that should be used when designing indications for use of leadless pacemakers. In the brief summary it is reported that the Panel agreed that indications for transvenous, single chamber (VVI) pacemakers apply to this class of devices and that guidance for certain population are already in place under the American Heart Association/American College of Cardiology/Heart Rhythm Association guidelines.
After the meeting, David Steinhaus, medical director for the Medtronic Cardiac Rhythm and Heart Failure Division, who presented to the FDA Panel on behalf of the company, commented: “We are committed to ensuring patient safety and device effectiveness over the long-term with ongoing monitoring of the Micra Transcatheter Pacing System (TPS) using a comprehensive post-approval study, a proprietary post-market surveillance programme and a rigorous implanter training programme, all of which are fully aligned with today’s panel discussions.”
In a press release, St Jude Medical stated that the FDA panel recommendations were consistent with many of St Jude Medical’s proposed recommendations related to the post approval study design and training for physicians. Mark Carlson, vice president of global clinical affairs and chief medical officer at St Jude Medical, who presented to the FDA Panel on behalf of the company, said: “Leadless pacing technology is a critical advancement for patients in need of a single-chamber ventricular pacemaker, and we are optimistic that this discussion is an important step toward offering patients leadless pacing technology in the United States.”
Both devices (Micra and Nanostim) have not been FDA-approved yet. Micra received CE mark approval in April 2015, based on the results from 60 patients implanted with the device for over three months in the Micra TPS Global Clinical Trial. The Nanostim received CE mark approval in August 2013, based on clinical results from 33 patients over three months in the LEADLESS study.
FDA’s Circulatory System Devices Panel of the Medical Devices Advisory
Voting members
David Yuh (Yale University, School of Medicine, New Haven, USA)
Richard Lange (University of Texas Health Science Center, San Antonio, USA)
David Naftel (University of Alabama at Birmingham, USA)
Joaquin Cigarroa (Oregon Health & Science University, Portland, USA)
David Kandzari (Piedmont Heart Institute, Atlanta, USA)
Temporary non-voting members included:
Jeffrey Brinker (The Johns Hopkins Hospital, Baltimore, USA)
David Slotwiner (School of Health Policy and Research, New York, USA)
Pamela Karasik (Veteran’s Administration Hospital, Washington, USA)
E Magnus Ohman (Duke University Medical Center, Durham, USA)
Jeffrey Borer (State University of New York, Downstate Medical Center, New York, USA)
Emily Zeitler (Duke University Hospital, Durham, USA)
Industry representative:
Naveen Thuramalla (Transonic Systems Incorporated, Ithaca, USA)
Consumer representative:
Naftali Frankel (Kew Gardens, USA)
Patient representative:
Debbie Dunn (Libertyville, USA)
Food and Drug Administration representatives:
Bram Zuckerman, director of the Division of Cardiovascular Devices
Dimitrus Culbreath, designated federal officer








