
Protaryx Medical has received 510(k) clearance from the US Food and Drug Administration (FDA) to market its proprietary transseptal puncture device.
According to the company, this milestone represents a significant advancement in its mission to redefine safe, efficient and reproducible left heart access for cardiac interventions, enabling broader adoption and improved delivery of minimally invasive therapies.
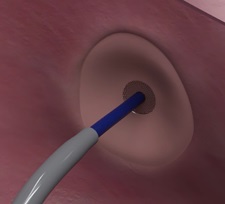
The device is a novel catheter system designed to improve the safety, accuracy and ease of transseptal puncture to facilitate access to the left atrium for procedures such as left atrial appendage closure (LAAC), transcatheter mitral valve procedures, and atrial fibrillation (AF) ablation. The device features an atraumatic, echogenic retractable nitinol mesh, as well as an extendable distal catheter, which are intended to enhance imaging visibility and aid deployment.
The device incorporates an echogenic, extendable atraumatic positioning probe and a standardised radiofrequency (RF) guidewire compatible with commercially available electrosurgical generators.
In an early first-in-human study (n=5), the device demonstrated procedural success in all cases with no reported device-related adverse events, minimal crossing time, and reduced fluoroscopic exposure.
“Receiving US FDA 510(k) clearance is a defining moment for Protaryx,” said David Mester, chief executive officer (CEO) of Protaryx. “This achievement reflects the dedication of our team to solving one of the most critical challenges in safe and efficient transseptal access procedures. We are now positioned to bring this transformative technology to physicians and patients across the USA.”
Protaryx co-founder James Gammie (Johns Hopkins Medicine, Baltimore, USA) added: “This clearance underscores the strength of the technology and its potential to set a new standard for transseptal puncture. By simplifying access to the left atrium, the device can expand procedural adoption while improving safety, precision, and ease of use.”







